►
From YouTube: Application of GPUs in Proteomics and Connectomics
Description
Fahad Saeed of FIU presents a talk on Application of GPUs in Proteomics and Connectomics. Pre-recorded session for GPUs for Science 2020 https://www.nersc.gov/users/training/gpus-for-science/gpus-for-science-2020/
A
So,
in
order
to
do
this,
we
use
multiple
modalities
from
different
ohmic
data
sets
that
come
from
different
kind
of
system,
biology
experiments,
and
then
we
develop
these
machine
learning.
Algorithms
that
allows
us
to
make
sense
of
these
data
sets
and
then
we
go
on
and
develop
high
performance
computing,
algorithms.
That
makes
this
process
more
scalable
right.
A
So
we
have
been
working
for
mainly
proteomic
data
sets
and
fmri
based
connectomic
data
sets
towards
this
end.
So
today
I
will
be
presenting
mostly
about
high
performance
algorithms,
that
we
have
been
developing
for
proteomics,
that
is,
mass
spectrometry,
based
proteomics
and
and
some
of
the
details
about
the
fmri
based
connectomix
and
I
will
be
happy
to
connect
offline
if
needed.
A
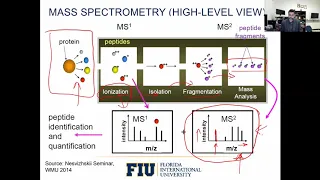
So
this
is
a
very
high
level
overview
of
mass
spectrometry
and
I
will
go
in
a
very
small
detail
of
how
the
data
from
mass
spectrometry
is
produced,
so
assume
that
these
are
the
proteins
that
are
that
we
will
try
to
study.
The
first
step
is
that
this
goes
into
the
ionization
stage,
where
all
of
these
specific
amino
acids
in
the
proteins
are
charged
at
a
very
high
are
charged
in
a
in
the
ionization
chamber.
A
And
then,
after
this,
the
ionization
chamber,
the
specific
peptides
go
into
the
isolation
chamber
depending
on
their
mass
right
and
once
they
are
in
the
isolation
chamber.
They
go
into
the
fragmentation
part
where
these
specific
small
molecules
peptides
are
then
bombarded
using
different
fragmentation
methodologies
right.
What
this
does
is
that
this
breaks
up
the
peptide
into
very,
very,
very
small
pieces
such
that
those
small
pieces
can
then
be
analyzed
using
their
mass
using
their
master
charge
ratio.
A
A
So
the
reason
that
we
consider
mass
spectrometry
based
proteomics
big
data
problem
is
primarily
because
of
two
reasons.
So
first
reason
is
that
the
mass
spectrometers
themselves,
the
instruments-
have
been
getting
more
and
more
efficient
right
and
they
have
been
getting
efficient
in
a
way
that
they
are,
they
have
been.
They
have
been
able
to
have
a
throughput
that
is
more
than
more
than
the
moore's
law.
A
There's.
Another
reason
why
we
considered
the
mass
spectrometry
based
proteomics
a
big
data
problem,
and
it
has
to
do
with
the
kind
of
computational
techniques
that
are
needed
to
process
the
mass
spectrometry
data
right.
So
here
I
will
show
a
very
high
level
schematic
of
why
this
is
the
case.
So
here
we
have
a
typical
mass
spectrum,
mass
spectrometers
right
that
have
produced
this
spectra,
and
these
spectra
can
be
anywhere
from
gigabyte
to
terabyte
level.
A
Okay
and
as
we
discussed
that
database
search
algorithms,
allow
us
to
process
this
data
using
databases
right,
so
the
databases
themselves,
the
protein
databases
that
are
used
to
that
are
used
as
a
reference
to
deduce.
The
peptides
are
rather
small
right,
so
there
can
be
hundreds
of
megabytes
right
and
because
of
this
there
are
many
people
who
might
say
that.
Well,
this
is
not
really
a
big
data
problem
right,
which
would
be
true,
but
the
problem
computational
problem
that
we
are
trying
to
solve
is
not
matching
this
spectra
to
this
specific
database.
A
If
you
closely
look
at
the
computational
problem,
computational
techniques,
especially
the
database
search
algorithms,
you
will
see
that
it
is
not
this
database
that
is
being
used
to
do
the
matching.
Rather,
this
database
is
then
expanded
into
a
much
larger
database
that
is
known
as
the
theoretical
species
specific
database,
where
the
database
is
expanded,
depending
on
the
parameters
that
are
being
given
to
the
search
algorithm
right.
A
So
in
this
specific
example,
you
will
see
that
here
we
have
this
data
set,
and
this
has
this
sequence
number
one
right,
and
for
this
specific
example,
we
assume
that
the
phosphorylation
is
being
requested
in
one
of
the
parameters
of
the
search
engine
right.
So
when
that
happens,
what
search
engine
does
is
that
it
takes
this
specific
sequence
and
then
goes
on
and
do
a
combinatorial
of
all
the
combinatorial
possibilities
that
might
be
associated
with
that
specific
sequence.
A
So
in
this
case,
you
have
this
sequence
and
all
of
these
sequences
are
done
expanded,
where
each
of
the
sequence
is
different
in
one
of
the
amino
acid.
So
so,
in
the
first
case,
the
phosphorylation
is
assumed
with
s.
In
the
second
case,
it
is
zoomed
with
t
in
the
third.
It
is
assumed
with
this
second
t
and
in
the
fourth
it
is
limited
with
the
y
and
so
on
and
so
forth.
A
Right-
and
this
is
just
one
example-
and
this
is
just
one
modification
that
is
being
requested
from
the
search
parameter
when
you
increase
the
number
of
modifications
in
the
parameters
for
the
search
engines.
The
common,
the
expansion
of
this
theoretical
database
is
rather
exponential
and
usually
people
do
their
search.
Engine
runs
using
two
or
three
post
translation
modifications,
just
because
the
results
do
not
get
scalable
with
increasing
size
of
increasing
number
of
parameters
right.
But
this
is
not
the
whole
story.
A
A
A
A
So
then,
the
the
way
that
the
these
search
engines
operate
is
that
they
assume
a
lot
of
filtering
mechanisms
that
allows
the
methods
to
be
scalable
right,
but
it
is
widely
known
that
those
filtering
mechanisms
lead
to
dark
data
in
proteomics,
which
means
that
it
leads
to
a
lot
of
data
that
might
not
be
seen
by
these
search
engines
right.
So,
in
order
to
solve
this
problem,
we
do
not
need
the
the.
A
So
this
is
what
the
schematic
of
the
our
gpu
based
algorithm
looks
like.
We
do
not
have
time
to
go
into
detail
for
all
of
that,
but
here
you
will
see
that
this
spectra
is
transferred
to
the
gpu
side,
where
only
the
intensity
arrays
are
transferred,
while
maintaining
the
mass
to
charge
ratio
spacing
of
the
of
the
of
the
spectra.
A
That
allows
us
to
go
on
and
process
the
data,
while
the
data
is
still
in
1d
array
right.
So
this
1d
array
is
then
processed
right
and
instead
of
transferring
all
of
the
data
back,
we
just
transferred
the
data
that
has
been
modified
because
of
this
reductive
algorithm,
and
that
saves
a
lot
of
bandwidth
in
from
going
from
gpu
to
cpu
right.
So
once
it
goes
back
once
this
data
goes
back
the
difference
of
difference
between
the
spectra,
and
that
is
process
and
spectra
that
has
changed.
A
A
So
then,
the
performance
that
we
got
for
the
execution
time
that
we
got
was
also
very
encouraging
for
these
data
sets
and
we
were
able
to
get
speed
ups
that
were
somewhere
between
100
and
400
times
as
compared
to
naive
approach.
That
only
gave
us
speedups
that
were
two
to
four
times.
For
this
large
mass
spectrometry
data
sets
so
most
of
the
work
here
on
gpu
based
mass
spectrometry
processing.
A
A
So
our
ongoing
efforts
is
towards
developing
a
few
of
the
fundamental
computational
motives
that
we
can
use
for
mass
spectrometry
based
proteomics,
and
there
are
specific
problems
that
we
are
working
on
related
to
load,
balancing,
localized
data
processing
and
making
sure
that
the
resources
on
these
large
supercomputers
are
used
in
an
efficient
manner,
and
we
run
all
of
our
code
on
exceed
supercomputers.
And
I'm
going
to
show
you
some
of
the
results
in
the
next
processing
that
we
have
been
doing.
A
The
basic
idea
is
that
we
want
to
be
able
to
process
these
mri
data
sets
in
a
very
fast
but
very
in
a
very
scalable,
but
in
a
very
accurate
manner,
and
we
are
able
to
do
the
and
and
the
and
the
data
that
you
get
from
these
mri
machines
are
rather
large
right.
So
our
our
effort
has
been
towards
developing
these
machine
learning
models
and
also
processing
these
machine
learning
models
using
gpus.
A
And
we
have
been
able
to
publish
that
in
a
frontiers
of
neuros
and
neuro
informatics
rather
recently,
and
the
the
reason
that
I
wanted
to
show
this
slide
is
that
gpu,
processing
or
high
performance
computing
for
scientific
data
sets
can
be
very
significant.
So
here
you
can
see
that
using
our
gpu
daemon
method,
which
is
a
generalized
method
that
can
be
used
by
different
people
who
might
not
be
very
familiar
with
cpg,
gpu
architecture
or
algorithms.
A
Just
using
that
method,
we
were
able
to
take
our
machine
learning
model
and
process
that
in
less
than
40
minutes
as
compared
to
other
methods
that
might
take
seven
hours.
So
a
a
carefully
designed
high
performance,
algorithm
4dbu
can
result
in
very
scalable
performances
for
many
of
these
scientific
data
sets,
which
is
which
is
very
useful,
especially
for
personalized
and
procedure
medicine.