►
From YouTube: 16 - Deep Learning for Quantum Chemistry - Justin Smith
Description
Deep Learning for Science School 2019 - Lawrence Berkeley National Lab
Agenda and talk slides are available at: https://dl4sci-school.lbl.gov/agenda
B
All
right,
so
this
is
a
this-
is
a
great
I'm
excited
to
give
a
talk
at
an
actual
machine
learning
conference
because
normally
I'm
talking
to
chemists,
and
so
they
don't
get
the
actual
machine
learning
aspect
of
things
that
we're
doing
so
are
actually
the
last
talk
was
Jennifer's.
Talk
was
really
good
because
it
kind
of
gives
you
a
idea
of
what
can
be
done
in
chemistry,
we're
coming
at
things
from
a
little
different
direction.
B
So
how
can
we
simulate
physics
of
atoms
using
machine
learning,
you're
interesting
cases
here
might
be
protein
simulation
simulation
of
liquids
or
the
simulation
of
materials
and
the
way
that
we've
traditionally
done
this?
We
have
some
energy
function,
potential
energy,
which
is
a
function
of
the
positions
of
the
atoms
in
the
system
and
your
electronic
structure
as
well.
If
you're
dealing
with
a
quantum
mechanics
problem,
we
calculate.
B
Oh
sorry,
we
can
get
forces
from
these
as
the
negative
gradient
of
that
energy
function,
and
these
are
guaranteed
to
be
conservative,
based
on
the
type
of
function
that
this
energy
function
is
and
we
carry
out
dynamics
using
Newtonian
physics.
It's
very
simple.
It
just
gives
you
a
kind
of
an
idea
of
what
one
of
these
simulations
looks
like,
and
actually
this
was
fully
carried
out
with
machine
learning,
we're
doing
these
types
of
simulations
right
now
at
Los,
Alamos,
National,
Lab.
B
We're
actually
scaling
these
up
to
millions
of
atoms.
At
this
point,
where
we're
doing
shock
simulations
through
materials
and
looking
at
how
the
wavefront
propagates
so
the
traditional
techniques
are
doing.
This
would
be
your
your
quantum
mechanics
methods,
which
we
briefly
had
mentioned
last
talk
where
you're
solving
the
time-independent
schrodinger
equation.
B
You
have
this
Hamiltonian
that
describes
different
different
physics
of
your
problem,
but
the
real
key
here
is
that
we're
treating
the
electrons
in
the
system
is
this
probabilistic
cloud,
and
so
we
have
this
very
expensive,
eigenvalue
problem
and
it's
an
iterative
eigenvalue
problem
that
we
end
up
having
to
carry
out
to
to
get
an
energy
out
of
this
problem.
There's
many
techniques
for
doing
this:
semi
empirical
methods,
density,
functional
Theory,
post,
our
Kukoc
and
the
order
and
the
number
of
electrons
and
your
system
grows
like
this.
B
So
this
is,
as
the
system
grows
in
size,
you
get
up
to
very
expensive
even
up
to
end
of
the
seventh.
Let's
hire
higher
cost
couple
cluster
methods.
Typically
in
organic
chemistry,
they
would
employ
these
force
fields
to
simulate
something
bigger.
Something
like
this
size
like
a
protein
in
a
binding
pocket
and
the
way
these
work
is
you
have
what
is
effectively
a
many-body
decomposition
of
your
system,
so
you
describe
torsional
four
body
interactions:
you'd
have
a
bonding
term.
B
Okay,
I'm,
not
gonna,
spend
much
time
here,
because
I
think
we've
seen
this
like
20
times
now.
But
the
idea
is
you,
we're
doing
supervised
learning
but
have
inputs.
We
have
some
function,
which
is
unknown
or
unknown
that
you're
putting
your
input
inputs
into
you,
get
a
label,
and
these
labels
are
the
things
you're
trying
to
fit
to.
B
So
it's
the
standard,
supervised
learning
process,
deep
neural
networks,
you
have
an
input
layer,
hidden
layers,
you
have
an
input
vector,
you
apply,
your
weights,
get
your
pre
activation,
apply
an
activation
function
and
you
get
your
output,
so
I
won't
spend
much
time
listening.
One
doesn't
know
how
that
works.
Yet
sorry,
so
we're
getting
there
for
our
application,
but
that
could
be
you
know,
depends
on
whatever
your
problem
is
it's
your
descriptor,
so
for
for
our
application,
what
we
end
up
inputting
is
effectively
coordinates.
B
B
The
problem
is,
our
function
is
very
slow
and
we
don't
want
to
have
to
run
molecular
dynamics
on
this
function
because
it
would
be
its
intractable
also
if
we
take
care
in
the
way
that
we
build
our
datasets
in
the
way
that
our
machine
learning
models
built
in
the
end,
we
can
just
train
two
small
sets
of
these
things
with
less
numbers
of
atoms
and
then
later
test
them
on
things
with
more
atoms.
This
is
this
idea
of
extensibility
extending
up
to
bigger
systems.
B
So
what
are
these
deep
learning
potential
as
well?
There's
a
couple
types
that
have
showed
up
in
literature
in
the
past,
so
this
type
here
you'd
have
some
kind
of
vector,
fixed
size,
vector
that
describes
just
a
molecule,
so
it
could
be
internal
coordinates.
What
that
means
is
distances
between
atoms
and
angles
between
them,
and
then
you
feed
this
into
a
deep
learning
model.
You
get
a
property
and
that's
it
very
simple.
The
what
we
believe
to
be
the
better
approach,
which
has
come
out,
probably
back
in
2005
issue.
B
Doesn't
seven
people
started
using
this,
for
you
have
a
fixed
size
vector
describing
the
chemical
environment
around
a
given
atom
so
for
three
atoms
in
this
water
molecule?
You
have
three
vectors
feed
this
through
your
deep
neural
network
model,
getting
a
tonic
property
sum
it
up,
and
then
your
total
property
represents
the
sum
of
individual
site
properties.
B
For
so
I
want
to
get
an
idea.
Give
you
an
idea
about
what
we
mean
when
we
talk
about
something
being
transferable,
or
so.
This
would
be,
like
general,
a
lot
of
people
talking
about
machine
learning
needing
to
generalize.
In
our
case
this
means
it
needs
to
be
transferable
and
extensible.
So
transferable
just
means
able
to
predict
on
things
not
in
the
training
set.
B
Extensible
is
subset
of
that
which
means
able
to
predict
on
things
larger
than
what's
in
your
training
data
set,
and
this
is
important,
because
we
can't
run
quantum
mechanics
to
get
information
to
get
data
about
a
protein
in
a
box
of
water.
That's
thirty!
Five
thousand
atoms
wouldn't
finish
in
my
lifetime.
If
I
try
to
run
this,
so
we
want
to
be
able
to
train
two
small
things
and
then
eventually
have
it
predict
on
these
bigger
systems.
B
So
the
problem
with
these
fixed
size
descriptors,
is
that
if
you
were
to
add,
say
another
molecule
to
the
system
and
have
a
water
dimer
that
fixed
size,
descriptors
no
longer
fixa,
as
you
now
have
more
bonds,
more
distances,
more
angles
between
atoms
and
so
this
grows.
And
then
your
deep
neural
network
needs
to
be
retrained.
B
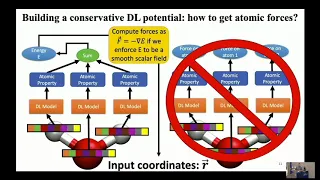
So
this
is
a
great
place
to
talk
about
again
putting
physics
back
into
your
model.
So
why
build
one
of
these
potentials?
It's
not
conservative.
We
know
from
physics
that
these
energy
function
should
be
conservative,
meaning
that
the
forces
should
conserve
energy
if
we
run
time-domain
simulations
on
them.
So
one
way
you
could
actually
predict
forces,
you
could
just
directly
predict
them
as
a
property
like
this.
B
The
problem
with
this
is
since
machine
learning
inherently
has
some
kind
of
error
to
it:
you're
never
going
to
conserve
forces
here,
because
you
have
some
error
preventing
these
things
from
summing
to
zero
and
it
actually
representing
a
conservative
vector
field.
The
better
way
to
do
it
is
to
have
your
model
predict
your
energy,
which
is
kind
of
the
definition
of
what
what
a
conservative
vector
field
is.
B
So,
to
represent
these
chemical
environments,
we
have
a
look
at
this
oxygen
here.
You
have
some
distance
an
atom
at
a
distance,
another
atom,
the
distance
and
angle.
If
you
have
another
molecule,
that's
outside
some
cutoff
radius
distance
away.
We
don't
consider
it
because,
typically
in
in
chemistry,
when
something
moves
far
enough
away,
the
interactions
drop
off
to
zero
fairly
quickly.
It's
not
always
the
case.
So
this
is
an
approximation.
B
B
So
there's
some
common
components
used
to
make
these
descriptors
invariant
back
in
2007,
Baylor
and
parinello
introduced
these
descriptors,
which
looks
something
like
this
here.
The
idea
is,
if
you
have
an
atom
centered
at
zero,
and
then
you
have
some
neighbor
J
at
3,
angstroms
away,
you're
peaking
these
different
functions
and
you're
reducing
overall
J
atoms
into
the
specter,
which
is
a
fixed
sized
script
vector.
So
this
now
is
a
once
you
sum
over
all
the
atoms.
This
describes,
basically
the
distribution
of
atoms
and
your
local
chemical
environment.
B
We've
released
modifications
disease
that
just
worked
better
in
certain
conditions.
This
one's
actually
cool,
who
says
these
mu
and
Sigma
or
learn
about
parameters
were
actually
having
the
machine.
Learning
model
learn
what
the
descriptors
should
look
like,
which
makes
a
little
more
sense.
We've
published
so
far.
Three
models
on
this
topic
so
originally
in
2017,
this
one,
which
was
one
of
the
first
that
showed
extensibility
and
transferability
within
an
entire
I,
would
say
an
entire
class
of
molecules,
organic
molecules.
B
So
if
we
train
this,
we
train
this
thing
to
about
22
million
different
DFT
calculations,
so
different
molecules
and
then
test
it
on
other
organic
molecules
and
we're
getting
really
good
performance
out
of
it.
These
models
bring
back
some
long
range
interactions
through
an
iterative
update
and
it
actually
acts
more
like
a
graph
convolution
neural
network.
So
we'll
talk
more
about
how
that
works
in
a
second
and
then
this
third
model,
which
I'll
also
talk
so
the
first
one
which
I
in
my
opinion,
is
the
simplest.
B
Is
you
have
these
radial
Raptors
that
build
Spectre,
which
I
was
talking
about
that
you're
summing
over
your
je
neighbors
and
you're
building
these
angular
descriptors,
which
sum
over
your
JK
neighbors
concatenating,
these
type
differentiate
them.
So
if,
like
your
neighbor
Jay's,
a
hydrogen,
you
just
put
that
in
a
specific
place
of
the
array,
your
neighbor
K
is
a
different
type.
You
put
in
a
different
type
of
the
array
you
sum
over
all
your
neighbors
feed
this
into
here.
B
It's
just
a
dense,
fully
connected
neural
networks,
the
most
basic
thing
ever
get
an
energy
prediction
sum
over
that
you
get
your
energy,
it's
a
very
simple
concept
and
then
for
forces.
Of
course
you
just
take
the
gradient
of
this.
Do
a
back
propagation
through
the
network
through
these
things
and
back
to
the
atomic
coordinates,
and
that's
what
gives
you
your
forces
on
atoms,
the
hip
and
instyle
neural
network,
which
works
more
like
a
graph
convolutional
model?
Basically,
you
just
take
this
radial
information.
B
You
build
your
radial,
descriptor
vector
and
then
here
you're
doing
this
interaction,
which
looks
like
a
graph
convolution
interaction
between
neighboring
sites
or
between
neighboring
neural
networks,
and
so
with
this
communication,
you
feed
this
into
a
dense
Network.
Actually,
these
are
these
are
recurrent
neural
Nets,
and
then
you
carry
out
this
interaction
iteratively
over
and
over
again.
B
Summing
up
your
predictions
along
the
way
until
your
end
result
is
an
energy
at
site,
I
summing
over
those
producers,
your
total
energy,
and
so
because
of
these
interaction
layers
being
iterative,
you're
actually
able
to
pull
in
longer
range
information
than
whatever
cutoff
you've
put
on
top
of
your
model
and
then
the
aim
net
style,
neural
network
potential
is
the
monster
of
these
things,
which
is
recently
accepted
in
science
and
dances.
So
you
build
your
radial
and
angular
descriptors.
B
You
combine
these
with
these
atomic
feature,
vectors,
which
are
now
learn
about
vectors
that
describe
the
type
of
atom
that
you
have
so
in
the
very
first
iteration
for
all
hydrogen's.
They
have
the
exact
same
vector
for
all
carbons.
In
the
exact
same
vector
you
combine
them
sum
over
all
J
neighbors
feed
it
through
a
neural
network.
You
get
an
update
to
your
a
vectors
so
now
in
the
next
iteration,
all
hydrogen's
are
not
equal
or
all
carbons
are
not
equal.
B
Oxygens
are
not
equal,
so
it's
like
learning
what
type
of
atom
this
should
be
described
as,
and
then
you
have
this
other
vector,
which
is
a
latent
space,
that
you're
learning
and
you're
concatenated
that,
with
the
new
atom
type
vector
this
gets
fed
through
the
atomic
neural
net,
another
atomic
neural
network.
So
this
is
on
each
site,
and
then
you
can
predict
different
quantities
from
here,
such
as
the
energy
at
that
site,
some
future
total
energy.
It
can
actually
predict
charges
as
well.
B
We
show
that
it
predicts
other
quantities
that
are
very
useful
to
chemists,
and
the
neat
thing
here
is
that
you,
through
this
process,
you
get
this
T.
These
iterative
updates
of
your
Tom
of
these
atomic
feature
vectors,
and
so
what
you
can
see
in
this
plot
appears
that
here
we
have
the
DFT
charge.
So
this
is
our
reference
level
of
theory
charge.
This
is
our
models
predicted
charge
on
iteration
1.
B
What
we're
looking
at
is
the
charge
on
this
sulphur
with
respect
to
changing
this
functional
group
are
with
these
different
groups
shown
in
the
plot,
and
the
key
here
is
that
these
are
all
outside
of
the
cutoff
radius
of
model.
The
model
can't
see
further
than
4.6
angstroms
explicitly,
but
through
this
iterative
update
at
T
equals
to
UX,
so
at
T
equals
1.
You
see
that
it's
flat.
It's
not
learning
anything
about
that.
Changing
that
R
group
and
a
further
cut
off
radius
and
then
at
later
iterations.
B
It
starts
matching
the
DFT
better,
because
it's
actually
able
to
pull
in
longer
range
information
from
the
chemical
environment,
and
so
to
give
you
an
idea
of
how
accurate
these
things
are.
We
actually
did
a
big,
active
learning
work
where
we,
where
we
let
the
model
drive
itself
through
chemical
space,
to
figure
out
what
new
data
should
be
included
on
the
next
iteration
and
our
models
shown
here
on
this
correlation
plot.
So
we
have.
This
is
our
model,
and
this
is
the
reference
level
of
theory,
its
density,
functional
theory,
and
this
is
a
log
plot.
B
So
actually,
the
vast
majority
of
the
data
is
pretty
spot-on.
Our
prediction,
these
two
DFT
BMP
m,
6
or
semi
empirical
methods
were
just
showing
us
a
baseline
to
show
you
that
even
comparing
quantum
mechanics
methods
themselves,
we
fit
very
accurate
accurately
to
our
level
of
theory.
These
are
faster
quantum
mechanics
methods
which
are
considered
less
accurate
but
they're.
What
people
typically
would
use
to
go
to
simulating
something?
That's
a
couple
hundred
atoms
and
sorry.
Oh,
this
is
a
test
set
yeah.
B
These
molecules,
every
one
of
them's,
bigger
than
anything
in
the
training
data
set
yeah.
So
this
is
this
is
a
protein.
That's
got
about
350
atoms,
this
one's
a
protein
with
some
212
atoms,
and
these
are
force
predictions
by
the
way,
not
direct
energy
predictions,
and
so
we
wanted
to
look
at
how
well
this
could
actually
be
used
in
a
you
know,
bulk
phase
simulation,
so
we
took
this
protein
with
a
drug
leggett
ligand
in
the
binding
pocket.
B
The
reason
this
is
interesting
as
drug
companies
would
be
interested
in
running
these
simulations
and
actually
obtaining
the
free
energy
of
this
thing
binding
inside
of
this
pocket,
and
then
they
can
get
an
idea
of
whether
or
not
it's
say
relatively
more
stable
than
another
molecule
that
they're
looking
at
in
this
system
at
about
35,000
atoms.
This
is
something
you
could
never
run
as
quantum
mechanics
ever
it
just
wouldn't
happen.
Your
you'd
run
out
of
memory
probably-
and
this
contains
seven
different
elements
inside
of
this
drug
ligand.
B
So
the
big-
the
big
thing
here
is
just
the
time,
is
amazing,
like
we're
able
to
get
about
a
time
step,
so
a
single
force,
calculation
for
all,
35,000
atoms
done
in
about
300,
milliseconds
or
so,
and
the
simulation
actually
converged
to
about
a
1
angstrom
rmsd,
which
means
that
the
structures
not
falling
apart.
So
it's
kind
of
showing
you
that
it's
actually
retaining
the
crystal
structure
that
we
originally
got
off
of
the
protein
databank.
B
Now,
the
really
cool
thing
about
this
is
imagine
you've
only
trained
a
model
to
organic
chemistry.
What
would
you
expect
it
would
do
if
you
then
showed
it
something?
That's
more
of
a
materials
problem
like
a
whole
bunch
of
carbon
in
a
box
at
a
high
temperature.
Most
people
say
it
wouldn't
work,
you'd
get
garbage
out
or
something,
but
actually
so
we
run
this
simulation.
It's
24
thousand
atoms,
2,500
Kelvin,
and
we
start
this
models.